Severity of Genotypes
Unfortunately, the most common AS genotype, deletion positive, tends to be the most severe, in terms of symptoms or characteristics.
Below is a an illustration Dr. Charles Williams used in his genetic presentation at the 2014 Canadian Angelman Syndrome Organization (CASS) Conference to show the relationship between AS genotypes and the severity of some AS characteristics.

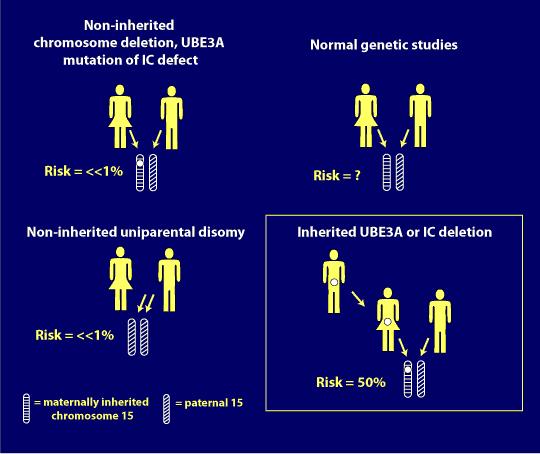
Risk of Recurrence in Angelman Syndrome
A genetic counselor can inform you on the possibility for Angelman syndrome to occur or recur through gathering family history and blood testing. The following information may be helpful in understanding the genetic risk of Angelman syndrome, but is not intended to replace genetic counseling.
1. Common chromosome deletion:
More that 98% of the chromosome deletion instances occur by a spontaneous event and thus they are not inherited; the recurrence risk is <<1% for these families. However, 1-2% of deletions occur because of an inherited abnormality in the maternal chromosome 15, such as a balanced chromosome translocation. Another very small group (e.g., only a few cases reported in the literature), can have AS due to a very small, maternally inherited chromosome deletion that involves a small area around and including the UBE3A gene. For these cases, the maternal recurrence risk is increased depending on the type of abnormality present. Chromosome study of the mother, including FISH, helps rule out inherited chromosome 15 abnormalities.
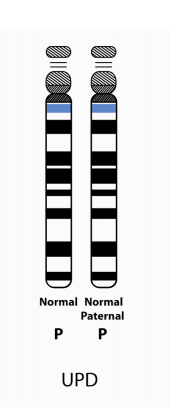
2. Paternal uniparental disomy (patUPD):
More than 99% of patUPD cases occur as an apparent spontaneous, non-inherited, event. If an individual has AS due to patUPD and has a normal karyotype, a chromosomal analysis of the mother should nevertheless be offered in order to exclude the rare possibility that a Robertsonian translocation or marker chromosome was a predisposing factor (e.g., via generation of maternal gamete that was nullisomic for chromosome 15, with subsequent post-zygotic “correction” to paternal disomy).
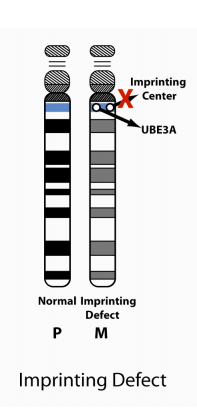
3. Imprinting Center (IC) Defect:
There are two types of IC defects: deletions and non-deletions. Non-deletion events do not appear to be inherited and have a <1% recurrence risk. Most deletions are not inherited but a significant proportion of them are (i.e., maternally inherited), and these confer a 50% risk for recurrence.
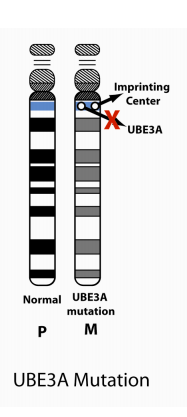
4. UBE3A mutations:
UBE3A mutation can either occur spontaneously (e.g., not inherited and with no increased recurrence risk) or be maternally inherited and have a 50% risk of recurrence (see below for imprinting inheritance).
5. Individuals with no known mechanism (all 4 above mechanisms have been eliminated):
For parents of AS individuals who have apparent normal genetic tests (no evidence for deletion, imprinting defect, UPD or UBE3A mutation), and thus their children are only clinically diagnosed, it is not known what the recurrence risk is. An increased risk seems likely but probably does not exceed 10%.
6. Germ cell mosaicism:
This term refers to a phenomenon in which a genetic defect is present in the cells of the gonad (ovary in the mother’s case) but not in other cells of the body. This occurrence can lead to errors in risk assessment because a genetic test, for example on a mother’s blood cells, will be normal when in fact a genetic defect is present in the germline cells of her ovary. Fortunately, germ cell mosaicism occurs very infrequently. Nevertheless, it has been observed in AS caused by the mechanisms of large chromosome deletion, Imprinting Center deletion and UBE3A mutation.
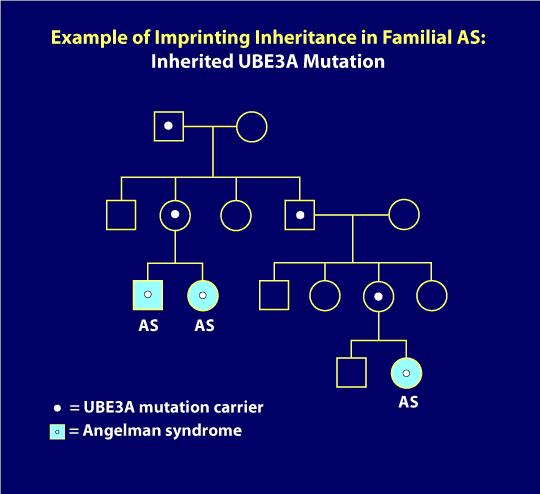
7. Imprinting inheritance:
UBE3A mutations and Imprinting Center deletions can exhibit imprinting inheritance wherein a carrier father can pass on the genetic defect to his children without it causing any problems, but whenever a female passes this same genetic defect on to her children, regardless of the sex of her child, that child will have AS. The pedigree diagram below illustrates imprinting inheritance. Here, AS has only occurred after a carrier mother passed on the gene defect (for example as in the two siblings with AS pictured on the left lower part of the pedigree). In addition, a distant cousin in this family also has AS due to the imprinting inheritance. In the diagram, individuals with the light blue circles or squares have AS but everyone else in the family is clinically normal. The white dots represent asymptomatic, normal carriers of the AS mutation. When an AS genetic mechanism is determined to be inherited, genetic testing of family members can usually identify carriers of the gene defect. As you might imagine, professional genetic counseling is advised in these situations.