Angelman syndrome (AS) is a rare neuro-genetic disorder that occurs in one in 15,000 live births or 500,000 people worldwide. It is caused by a loss of function of the UBE3A gene in the 15th chromosome derived from the mother.
Angelman syndrome shares symptoms and characteristics with other disorders including autism, cerebral palsy and Prader-Willi syndrome. Due to the common characteristics, misdiagnosis occurs often.
People with Angelman syndrome have developmental problems that become noticeable by the age of 6 – 12 months. Other common signs and symptoms usually appear in early childhood like walking and balance disorders, gastrointestinal issues, seizures and little to no speech. Despite these symptoms, people with Angelman syndrome have an overall happy and excitable demeanor. An individual with AS will light up a room with their smile and laughter.

When families first learn about Angelman syndrome, they often hear a list of things their child may never do. But that information doesn’t tell the whole story.
Every individual with Angelman syndrome has their own pace and personality, but there are so many beautiful things that they CAN do.
Visit our Wall of Accomplishments to see examples of progress and possibilities! Individuals with AS walking, swimming, climbing and exhibiting independence! Many of these individuals’ parents were told their child would never do these things.
Each example tells a story of resilience, determination, and hope. These accomplishments are reminders that with the right support, individuals with Angelman syndrome will continue to surprise, inspire, and achieve.



Angelman syndrome was first identified by Dr. Harry Angelman, an English physician at Warrington General Hospital.
Dr. Angelman first observed three children who were unrelated but showed similar symptoms of severe intellectual delay, stiff, jerky gait, lack of speech, seizures, motor disorders and a happy demeanor.
Then, while vacationing in Italy, Dr. Angelman observed an oil painting called… A Boy with a Puppet by the renaissance artist Giovanni Francesco Caroto at the Castelvecchio museum in Verona. Reminded of the children, Dr. Angelman published a paper in 1965 that described what he called “puppet children”.
It wasn’t until 1982, when Dr. Charles Williams and Dr. Jaime Frias of the department of Pediatrics, Division of Genetics, University of Florida College of Medicine, Gainesville submitted a paper to the American Journal of Medical Genetics. The paper reported studies of six patients and compared their data to those from previous reports. It sighted – severe developmental delay, “puppet-like” gait, craniofacial abnormalities, and frequent episodes of laughter. It became clear the syndrome was more common than previously thought. They proposed the name of this disorder be changed to Angelman Syndrome.
In 1986, Dr. Williams started the Angelman Research Group (ARG) for facilitating research and education of Angelman syndrome. A few years later, in 1990, the ARG became the Angelman Syndrome Foundation. See more about the history of the ASF.
Some symptoms can vary and be more severe than others, but in most children diagnosed with AS, the following are present:
These can vary from individual to individual, but common delays are:
Infants (0-24 months): inability to support one’s head, pull oneself up to stand and delayed motor skills like crawling.
Feeding issues due to problems sucking or swallowing.
Young children: Delayed ability to walk and an unstable gait or balance issues.
Seizures are a common symptom of Angelman syndrome, affecting the majority of individuals with the condition. They typically begin between 18 months – 3 years old and can vary in type and severity.
A consistently happy demeanor is one of the most recognizable traits of Angelman syndrome. Individuals with often exhibit frequent smiling, laughter, and an easily excitable disposition.
Sleep disturbances are a common trait of Angelman syndrome, beginning at a young age. Individuals with may have trouble falling asleep, staying asleep, or may sleep for shorter durations than typical.
Sleep issues can improve with age. Many families rely on a combination of behavioral strategies and medical interventions to support better rest.
Infants display lack of cooing or babbling; young children usually use nonverbal methods of communication because conversational speech is either absent or limited to very few words.
Angelman syndrome (AS) is caused by decreased or absent function of the UBE3A gene. It’s important to keep in mind that in typical humans, the UBE3A gene from our father is silent and the brain uses the UBE3A gene from our mother during development.
Humans have 46 chromosomes inside every cell in their body. We receive 23 chromosomes from our mother and 23 from our father. Different genes are located in each chromosome. The Angelman syndrome gene, UBE3A, is located at chromosome 15.
Some genes on the chromosome are turned on or expressed and others are turned off or silent. In typical humans, the UBE3A gene from our father is silent and the UBE3A gene from our mother helps our brain develop. However, in individuals with Angelman syndrome, there is a problem with the UBE3A gene from the mother and the brain cannot get the information it needs to develop and control speech, movement and learning. All of this action in the chromosomes takes place during fetal development, and thus is part of a person’s genetic makeup.
This image portrays a typical human with 2 number 15 chromosomes, one from the mother (M, maternal) that is expressed, and one from the father (P, paternal) that is silent.
When the UBE3A gene does not function normally, the individual has Angelman syndrome. Scientists around the world, are studying the UBE3A gene and trying to find ways to turn on or unsilence the copy from the father.
There are 4 ways that Angelman syndrome can occur. These are called genotypes. Each genotype has a different mechanism that results in AS.
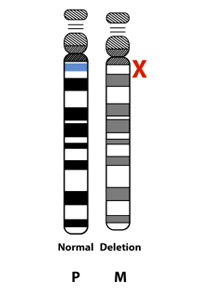
The most common cause of AS is known as “deletion positive” or “deletion.” People with deletion positive AS have a deletion of the maternal UBE3A gene and other nearby genes. About 70 percent of people with AS have the deletion subtype.
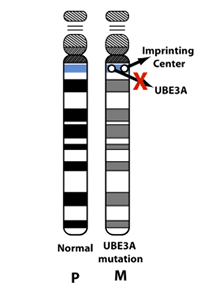
AS can be caused by a misspelling or error within the maternal copy of the UBE3A gene. Depending on the specific error in the UBE3A gene, there may be reduced or no function of the maternal UBE3A gene in the nervous system. Some people with mutation type AS may have a small deletion that only impacts the UBE3A gene. The “mutation” subtype makes up ~13 percent of cases of AS.
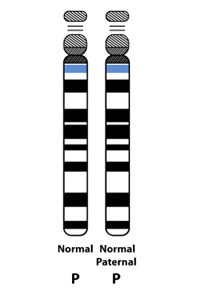
Typically, people inherit one half of their genetic material from each parent. However, it is possible to inherit two copies of genetic material from the same parent (“uniparental disomy”). If a person inherits two copies of the UBE3A gene from their father (“paternal uniparental disomy), there will be no expression of maternal UBE3A in the nervous system causing the symptoms of AS. Paternal UPD causes ~10 percent of cases of AS.
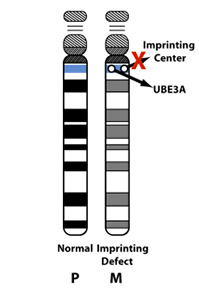
There are genetic control centers (“imprinting centers”) that direct genes to be turned on or off. If there is an error in the imprinting center, this can cause the maternal UBE3A to be silenced inappropriately. About ~7 percent of cases of AS are caused by imprinting center defects.
Unfortunately, the most common AS genotype, deletion positive, tends to be the most severe, in terms of symptoms or characteristics.
Below is a an illustration Dr. Charles Williams used in his genetic presentation at the 2014 Canadian Angelman Syndrome Organization (CASS) Conference to show the relationship between AS genotypes and the severity of some AS characteristics.
Due to common characteristics that AS shares with other disorders (developmental delays, motor issues, and lack of cooing, babbling or speech), 50% of individuals with Angelman syndrome are originally misdiagnosed. Late or misdiagnosis may cause individuals to lose opportunities for early intervention programs, resources, personalized support or life-changing treatments.
Testing and diagnosis of AS is done through a medical doctor. To find a clinician in your area to perform genetic testing, see the Genetic Testing Registry or contact the ASF.
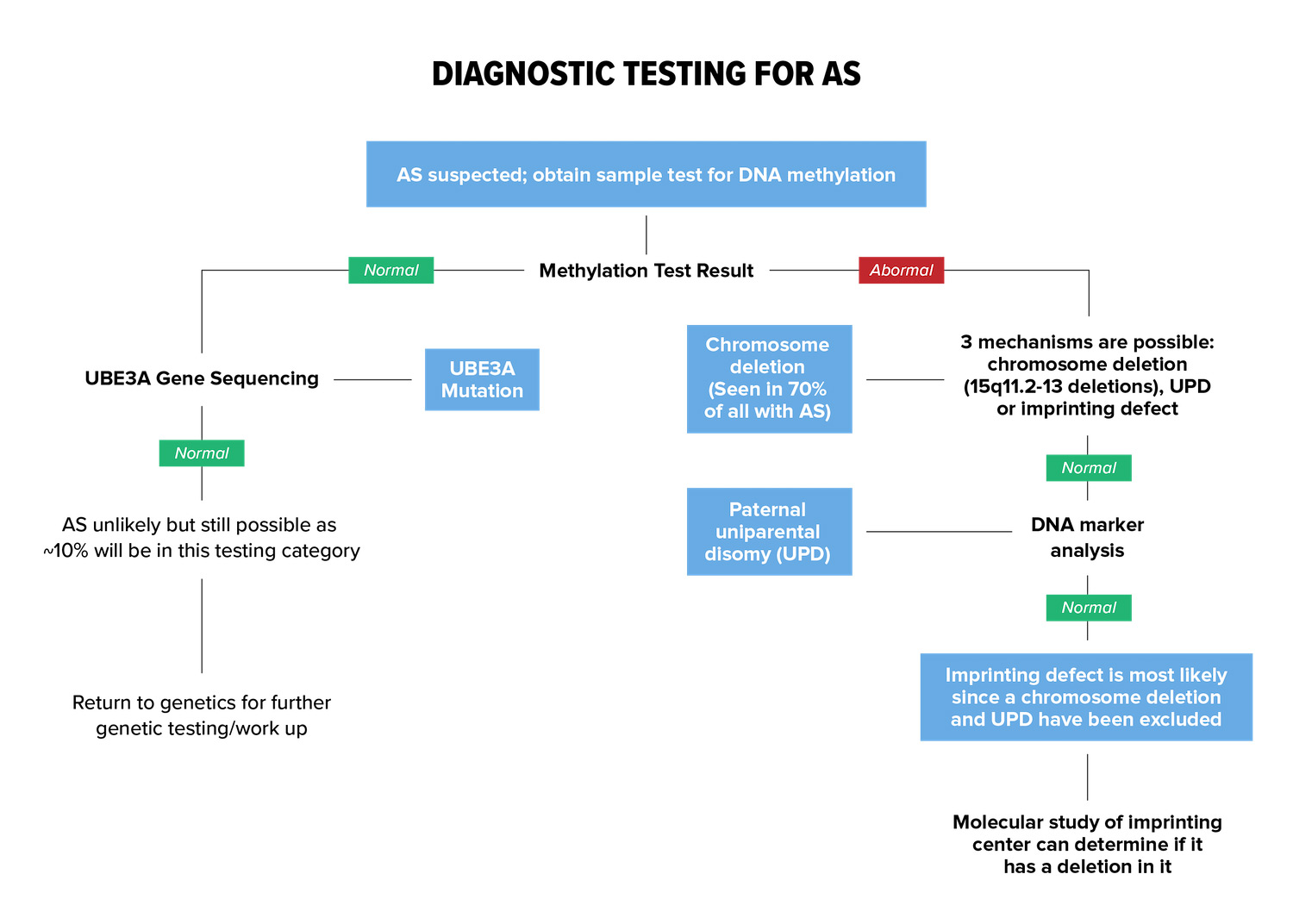
Below are the tests that help determine whether AS is present. All of these tests are done through a blood test and the final results of genetic testing can take a couple weeks or more.
A methylation test can identify whether the UBE3A gene is functioning properly. If the methylation test determines that UBE3A is abnormal, that means the person has AS and the next step is to determine the genetic type.
If the methylation test is normal, DNA sequencing of the UBE3A gene is performed to confirm whether the UBE3A gene is functional. In this case it is likely that the patient has a condition that mimics AS.
A FISH 15 test (fluorescent in situ hybridization) or comparative genomic hybridization (CGH) test will find chromosome deletion (or deletion positive), which is 70% of all individuals with AS. If the FISH 15/CHG test is normal, then the person is not deletion positive.
DNA Marker Analysis will determine whether the type of AS is Paternal uniparental disomy (UPD). If the type is not UPD, then an Imprinting Center Defect is assumed, but molecular study of the imprinting center can confirm.
After receiving a diagnosis of Angelman syndrome, many parents are overwhelmed by the fact that they know nothing about the syndrome and the road ahead.
The good news is that we know that an individual with AS is able to do and accomplish more than was believed years ago. Parents were told their child would never walk and never communicate. We know now, that is not true. People with AS today are walking, communicating and graduating high school.
Early intervention is key. Talk to your child’s doctor about potential therapies to help with development. Will there be some rough road ahead? Probably. But the Angelman Syndrome Foundation is here to help as well as a community of families, caregivers, therapists, researchers and physicians from all over the world.
If your child was recently diagnosed with Angelman syndrome, see the Newly Diagnosed page and fill out the form today.
We highly recommend visiting a clinic in the ASF Clinical Network to establish or supplement the care of individuals with Angelman syndrome.
A variety of treatments can help manage symptoms and improve the quality of life for individuals with Angelman syndrome. This requires a multidisciplinary approach involving a team of specialists.
Seizure Management
Seizures are a common symptom of Angelman syndrome, often beginning between 18 months-3 years of age.
Anti-seizure medications (anticonvulsants) are typically used to manage and reduce the frequency of seizures. Regular consultations with a neurologist are essential for monitoring and adjusting medications.
Behavioral Interventions
Behavioral therapies can help address some of the social and emotional challenges associated with Angelman syndrome.
Applied Behavior Analysis (ABA) and other behavioral strategies may be employed to improve sleep patterns, manage hyperactivity, and reduce anxiety.
Sleep Support
Most individuals with AS will have problems with sleep. Some common sleep issues in AS include difficulty settling, shorter sleep duration, and more fragmented sleep.
It is important to assess for health problems that may impact sleep, consider behavioral strategies to improve sleep, and to work with neurology and/or a sleep specialist if needed.
Therapies to Support Development
Speech Therapy: Due to lack of verbal speech, individuals may benefit from alternative communication methods, like sign language or augmentative and alternative communication (AAC) devices.
Physical Therapy: Helps improve balance, coordination, and overall motor skills, which are often delayed in individuals with AS.
Occupational Therapy: Focuses on improving the ability to perform daily activities such as feeding, dressing, and mobility.
Gastrointestinal Health
Many individuals with AS experience digestive concerns, such as feeding issues, constipation or acid reflux, which can impact their comfort and overall well-being. Gastroenterologists and Diet Specialists can recommend dietary changes and supplements to help alleviate these issues.
Vision Health
Strabismus and Cortical Visual Impairment (CVI) are possible issues that affect the eyes. Regular check-ups allow eye care specialists to catch any issues early and provide timely intervention to support your loved one’s development and interaction with the world.
There are numerous additional treatment options and resources that can further support individuals with Angelman syndrome, each aimed at fostering development and maximizing independence based on specific needs.
Find resources to share with your medical team in the Find Resources section, select Medical.
Research into potential treatments for Angelman syndrome is ongoing and there are clinical trials happening to test the efficacy and safety of drug therapies. The Angelman Syndrome Foundation actively supports research initiatives that may offer new treatment approaches and hope for the future.
Some symptoms of Angelman syndrome improve as individuals get older. Sleep issues and seizures tend to become less severe or infrequent. Because of mobility issues, obesity and scoliosis can develop in adolescence.
The life expectancy of people with Angelman syndrome is normal. Angelman syndrome itself does not cause death. However, there can be severe complications due to some of the symptoms of the syndrome, such as seizures and aspiration pneumonia. There is also the possibility of accidents due to walking and balance issues and attraction to water that can cause severe injury.
Individuals with AS will require life-long care, but can live long, happy lives.
Find out more about the Natural History Study that is building a better understanding of AS through a lifetime.
If you have a family history of AS, a genetic counselor can help assess the chances to have a child with AS (“recurrence risk”). It is important to share any genetic testing results with the genetic counselor for the most accurate assessment. The following information is not intended to replace genetic counseling.
In families with deletion positive AS, typically the chance to have another child with deletion type AS is less than 1 percent. Most cases of deletion positive AS happen by chance, and are not inherited. However, 1-2 percent of deletions occur because of a rearrangement of genetic material (a “balanced chromosome translocation”) in the mother. Another very small group (e.g., only a few cases reported in the literature), can have AS due to a very small, maternally inherited deletion that involves a small area around the UBE3A. Chromosome study of the mother, including FISH, helps rule out inherited chromosome 15 abnormalities.
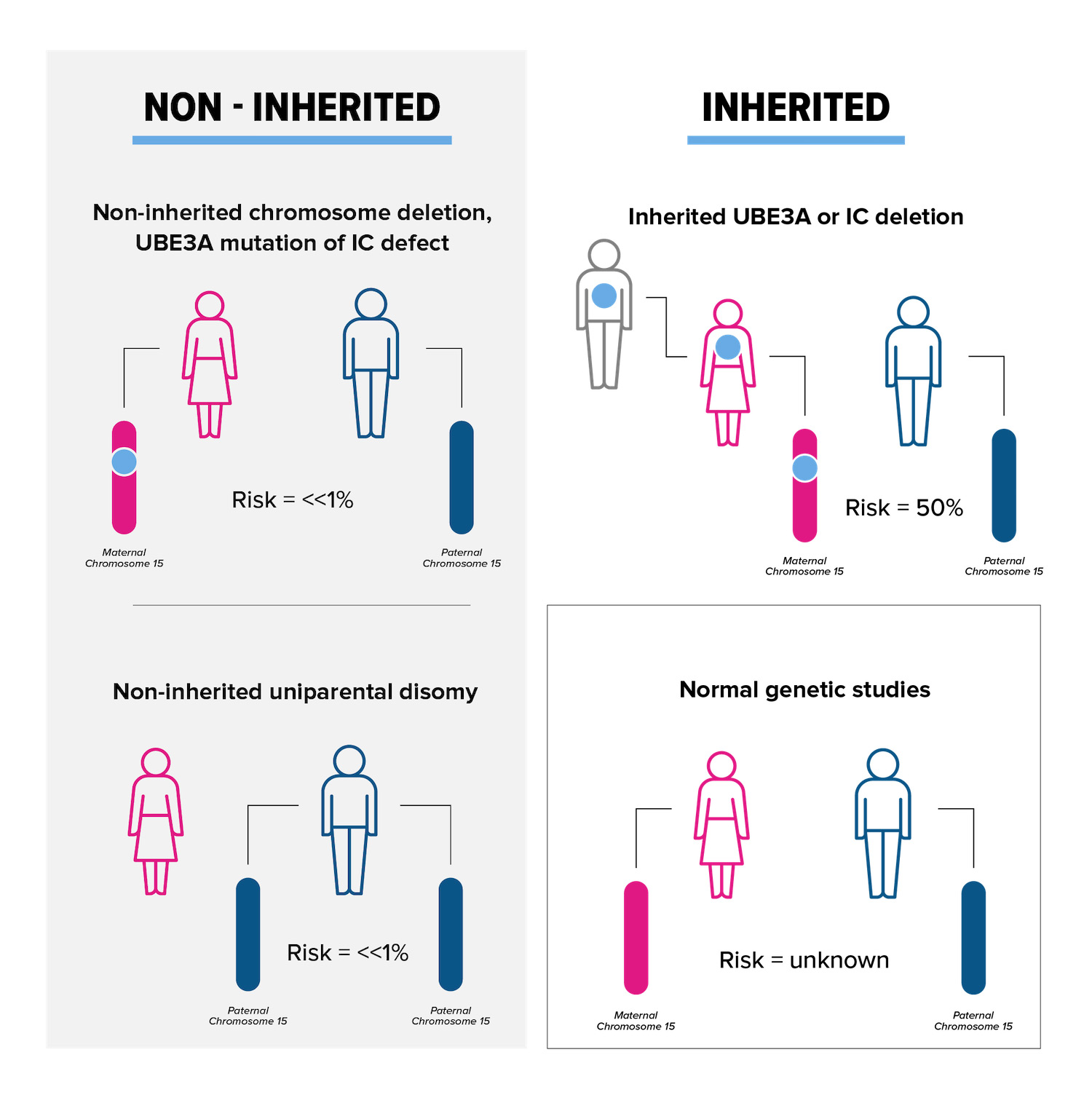
The UPD subtype of AS is not inherited in more than 99 percent of cases. However, chromosome studies should be offered to the mother to be sure that there is not a chromosome rearrangement predisposing to the UPD subtype.
Imprinting center defects (ICD) can happen by chance or be inherited from the mother. If the imprinting center defect was inherited from the mother, there may be as high as a 50 percent chance to have another child with ICD type AS. If the ICD was not inherited, the recurrence risk is less than 1 percent.
UBE3A mutations can happen by chance or be inherited from the mother. Similar to to the ICD subtype, if the mutation is inherited from the mother, there may be as high as a 50 percent chance to have another child with UBE3A mutation type AS. If the UBE3A mutation was not inherited, the chance to have a second child with AS is likely less than 1 percent.
For parents of AS individuals who have apparent normal genetic tests (no evidence for deletion, imprinting defect, UPD or UBE3A mutation), and thus their children are only clinically diagnosed, it is not known what the recurrence risk is. An increased risk seems likely but probably does not exceed 10%.
In very rare cases, it is possible for a mother to have a genetic change for AS only in her reproductive cells. This has been reported in families with deletion positive AS, ICD subtype, and UBE3A mutation subtype.
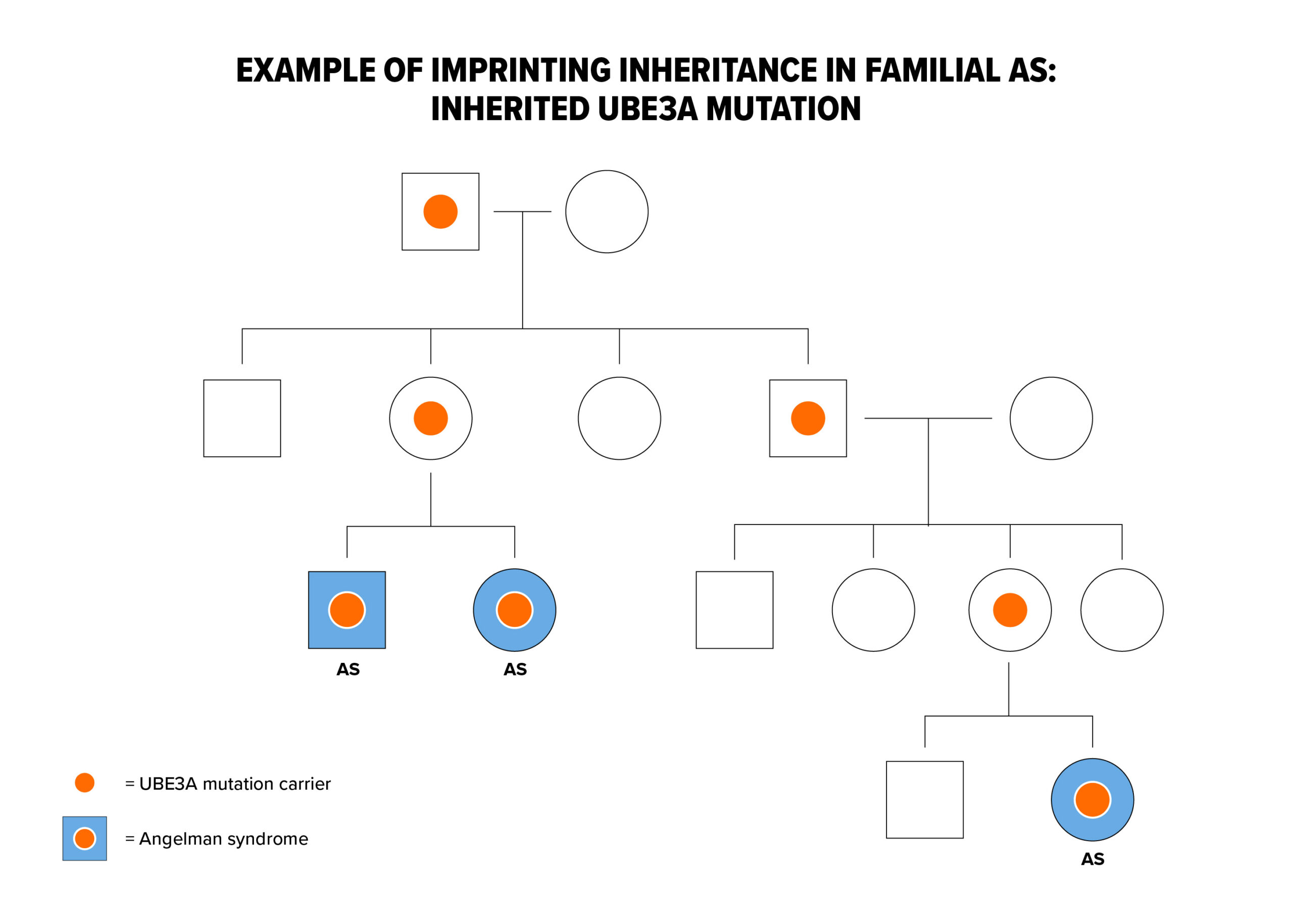
UBE3A mutations and Imprinting Center deletions can exhibit imprinting inheritance where a carrier father can pass on the genetic defect to his children without it causing any problems, but whenever a female passes this same genetic defect on to her children, regardless of the sex of her child, that child will have AS. The diagram illustrates imprinting inheritance.
Here, AS has only occurred after a carrier mother passed on the gene defect (for example as in the two siblings with AS pictured on the left lower part of the diagram). In addition, a distant cousin in this family also has AS due to the imprinting inheritance. In the diagram, individuals with the light blue circles or squares have AS but everyone else in the family is clinically normal. The white dots represent asymptomatic, normal carriers of the AS mutation. When an AS genetic mechanism is determined to be inherited, genetic testing of family members can usually identify carriers of the gene defect. As you might imagine, professional genetic counseling is advised in these situations.
It is believed that there is a high chance of finding a cure for Angelman syndrome, due to the fact that scientists know what causes AS and have been able to reverse it in mouse models.
Watch Dr. Arthur Beaudet discuss a cure for AS:
